Biology Forum › Molecular Biology › problem with cloning using pTYB2
- AuthorPosts
- May 5, 2008 at 8:57 pm #9591
 ripplemmParticipant
ripplemmParticipantI am trying to clone a 300 bp gene into pTYB2 vector (7.4kb)for later protein purification method by IMPACT-CN. NdeI (producing sticky end) and SmaI (blunt end) restriction sites were constructed into primers corresponding to the N- and C- termini of the gene and incorporated by PCR amplification. The PCR product and the vector were both double digested with SmaI and NdeI respectively. After gel purification, the digested insert (gene) and the vector were used to perform ligation with T4 DNA ligase. Since the expected product is not very different from the digested vector, it was really hard to tell whether the desired product was present on the gel. Then the ligation mixture was used to transform to JM109 competent cells. NO COLONIES WERE GROWING. 😥 I have done a bunch of controls. It seems that I have issue with either ligation or transformation, or both. I really hope someone here can help me with this. Molecular biology is all about experiences, great care and luck.
Here are the procedures.
(1). Double digestion
2 ug DNA
SmaI 25 degree C digestion for 2.5 hours
increase the temperature to 37 degree C, add NdeI, continue the digestion for 2.5 hours
deactivate the enzymes by heating at 65 degree C for 20 min
(2). Gel purification of digested products
phenol/chloroform extraction (to remove the enzyme)–>EtOH precipitation (with NaOAc added)–>spin down the DNA–>70% EtOH cold wash–>dry in SpeedVac–>resuspend in 20 uL of TE–>add agarose dye,load on 1% agarose gel–>cut the band under UV light–>electroelution using dialysis tubing–>EtOH precipitation70% EtOH cold wash–>dry in SpeedVac–>resuspend in 20 uL of TE
(3). Run a 1% agarose gel with known concentration sample to estimate the concentration of insert and vector
(4). Ligation with T4 DNA ligase @16 degree C overnight (since I have both sticky and blunt end)
vector:insert, I have tried 1:5 and 1:10, reaction volume is 30 uL.
(5).Transformation
positive control : uncut vetor only, it works really well
negative control: no colonies
experiment: 2 uL of ligation mixture into 100 uL of JM109 competent cells, electroporation–>1 mL SOC medium–>37 degree C 1 hour–>spread 200 uL culture on LB/Amp plates
NO COLONIES at allHere are the controls I have performed.
(1). Long time/sequential digestion
I tried single digestion with SmaI first (overnight),P/C purifying digested product, then digest with NdeI (overnight). Then self ligation was performed. Apparently, there is no difference between short-time digestion and long-time digestion.
(2). Self ligation control
vector: ladder has been observed
insert: ladder has been observed, which includes monomer, dimer, trimer…This implicates both NdeI and SmaI worked, otherwise I should see only monomer and dimer if only one enzyme worked.
(3). Uncut vector was able to transform to JM109 pretty well.Could be my insert toxic to the cell?
Any comments are welcomed! - May 6, 2008 at 5:08 pm #83936blcr11Participant
I don’t have any direct experience with the intein system. I have read about the system a bit. Someone else on the site was having trouble with it too. Read down through this rather lengthly thread. The intein questions are near the end of it:
http://biology-online.org/biology-forum/about12887.html
Your protocol looks OK to me. Are you sure your pcr product successfully digested with both enzymes? You’ve designed your pcr primers to contain 6-8 extra bases beyond the restriction site, I assume. The junk sequence is only there to help with digestion. It serves no other purpose, but if it isn’t there some REs won’t cut very well near the ends of linear DNA.
I think the ligation didn’t work, and my first guess would be that the pcr product didn’t digest with both enzymes–but that is just a guess. Your ligation is going to be a mixed sticky-end and blunt-end ligation. You could try doing a short (2-8 hr) ligation at 30 ul (which I guess is already more dilute than the "standard" ligation mix, so maybe what I’m suggesting is stupid, but when did that ever stop me) and then adding more enzyme and buffer to 50 or even 75 ul. The thinking is to favor the easy-to-do sticky-end ligation for a short while at higher concentration of ends to favor intermolecular ligation, but then dilute these once-ligated things to favor intramolecular blunt-end ligation at the SmaI site. Don’t dilute the enzyme. You want to keep the ligase concentration the same, or even a little higher while you dilute the partially ligated vector + insert.
You can (well, you can if you ever get colonies) screen for insert by colony pcr, which will be more sensitive than restriction digestions and saves you the trouble of having to make mini/maxi prep DNAs before you know whether or not there is anything worth looking at.
- May 8, 2008 at 12:25 am #83971ripplemmParticipant
Thanks a lot blcr11!
I actually found that post by googling for pTYB2. Because it seemed that I have different issues, I thought it would be a good idea to ask for suggestions here.To answer your questions,
(1). My PCR product should be double digested well. I drew my conclusion based on the self-ligation results. The double-digested PCR product was able to form dimer, trimer,tetramer,even pentamer. If only one enzyme worked, only monomer and dimer would be observed.(2). The ligation reaction, the total volume of 30 uL was used according to the protocol from ROCHE because we are using T4 DNA ligase from ROCHE.
DNA: 0.8 ug total (vector and insert)
10X ligation buffer: 3 uL (ATP included)
H2O:30-other components’ volume
T4 DNA ligase: 2 uL (total 2 U)(3). I did not see any colonies on my plates before. The day before yesterday I did another transformation with top 10 competent cells. SURPRISINGLY, there were tons of cells growing on the plates.
+/- controls were fine
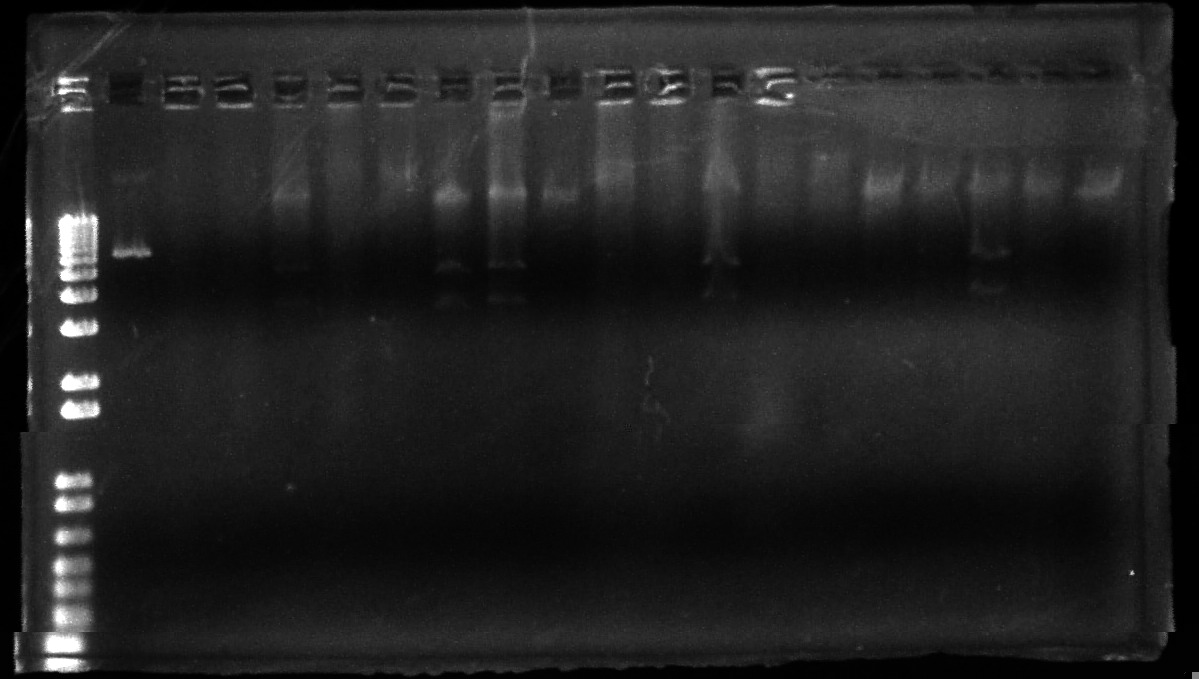
I was really excited, then I did 5 mL culture plasmid miniprep (PROMEGA kit) to screen 10 colonies. I have to say the cells were overgrown because I did not know top 10 grows much faster than JM109. After 5 hours growing, the cell density was really high. I did the minipreps last night. This morning, I ran a gel to check whether I got the expected plasmid. HOWEVER, the weirdest thing happened. I saw two bands for each sample, except the genomic DNA. These two are >= 1kb different so they could not be super coil and open circular. I have never seen this phenomenon before. I attach the pictures here, hopefully somebody has seen this before.from left to the right
1kb+ ladder
uncut vector (7.4kb)
miniprep colonies (the expected new plasmid should be 7.7 kb)Attachments:

- May 8, 2008 at 12:42 am #83972ripplemmParticipant
There is something I did not say clearly in my first post. I am doing cloning for two different proteins separately. One is 300 bp, the other is 1 kb. pTYB2 is 7.4 kb.
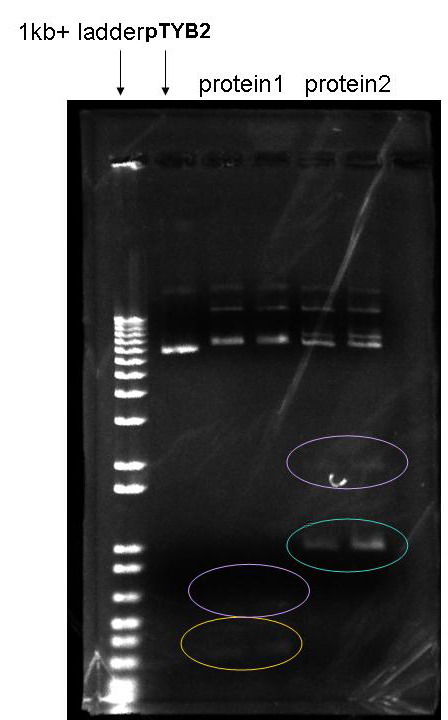
Here is a picture for my ligation reactions.
pTYB2 is the original vector, I was using it as a marker on this gel.
from left to the right
Lane content
1 1kb+ ladder
2 pTYB2
3 ligation (double digested pTYB2+double digested PCR product of protein1 gene) 1:5 ratio
4 same as lane3 but 1:10 ratio
5 ligation (double digested pTYB2+double digested PCR product of protein1 gene)
6 same as lane5 but 1:10 ratiothe yellow circle: 300 bp insert for protein1 gene
the blue circle: 1 kb insert for protein2 gene
the purple circle: dimers for insertsAttachments:
- AuthorPosts
You must be logged in to reply to this topic.